Was ist GMP?
Unter GMP (aus dem Englischen „Good Manufacturing Practice“ = Gute Herstellungspraxis) versteht man den Teil der Qualitätssicherung, welcher gleichbleibende Qualitätsstandards bei der Produktion und Prüfung von Arzneimitteln oder Wirkstoffen sicherstellt.
Dies ist ein kurzer Ausschnitt aus dem eLearning Kurs zu GMP Grundlagen

Die menschliche Gesundheit steht dabei im Vordergrund. Im Alltag müssen sich Patient:innen auf die hohe Qualität eines Medikamentes verlassen können, da sie diese nicht selbst überprüfen können. Die Arzneimittelqualität wird vor allem durch folgende Faktoren bestimmt:
- Identität (sind die vorgegebenen Inhaltsstoffe enthalten?)
- Gehalt (sind die Inhaltsstoffe in der vorgegebenen Menge vorhanden?)
- Reinheit (ist das Medikament frei von Inhaltsstoffen, die nicht hineingehören?)
Das deutsche Arzneimittelgesetz (AMG) fordert in §1, dass die Qualität, die Wirksamkeit und die Unbedenklichkeit einer Arznei sichergestellt werden müssen. Diese sind der zuständigen Behörde nachzuweisen, welche die Zulassung für das Medikament erteilt, denn: ohne Zulassung darf ein Medikament nicht an Patientinnen oder Patienten vertrieben werden. Wie lassen sich diese Ansprüche erfüllen? Und: wie lässt sich eine gleichbleibende Qualität sicherstellen, wenn nicht etwa jede gepresste Tablette einzeln geprüft werden kann? Dazu dient die gute Herstellungspraxis.
Anzuwenden sind deren Prinzipien dabei in fast allen Unternehmensbereichen, von der Forschung und Entwicklung über Produktion, Qualitätskontrolle und Lagerung bis hin zum Vertrieb. Wer Arzneimittel oder Wirkstoffe gewerbsmäßig herstellt, prüft, lagert oder in den Verkehr bringt, muss demnach eine Reihe von definierten Anforderungen umsetzen. Der Grundsatz lautet: Unternehmen müssen Medikamente so herstellen, dass sie für ihren vorgesehenen Gebrauch geeignet sind.
Woher kommt GMP?
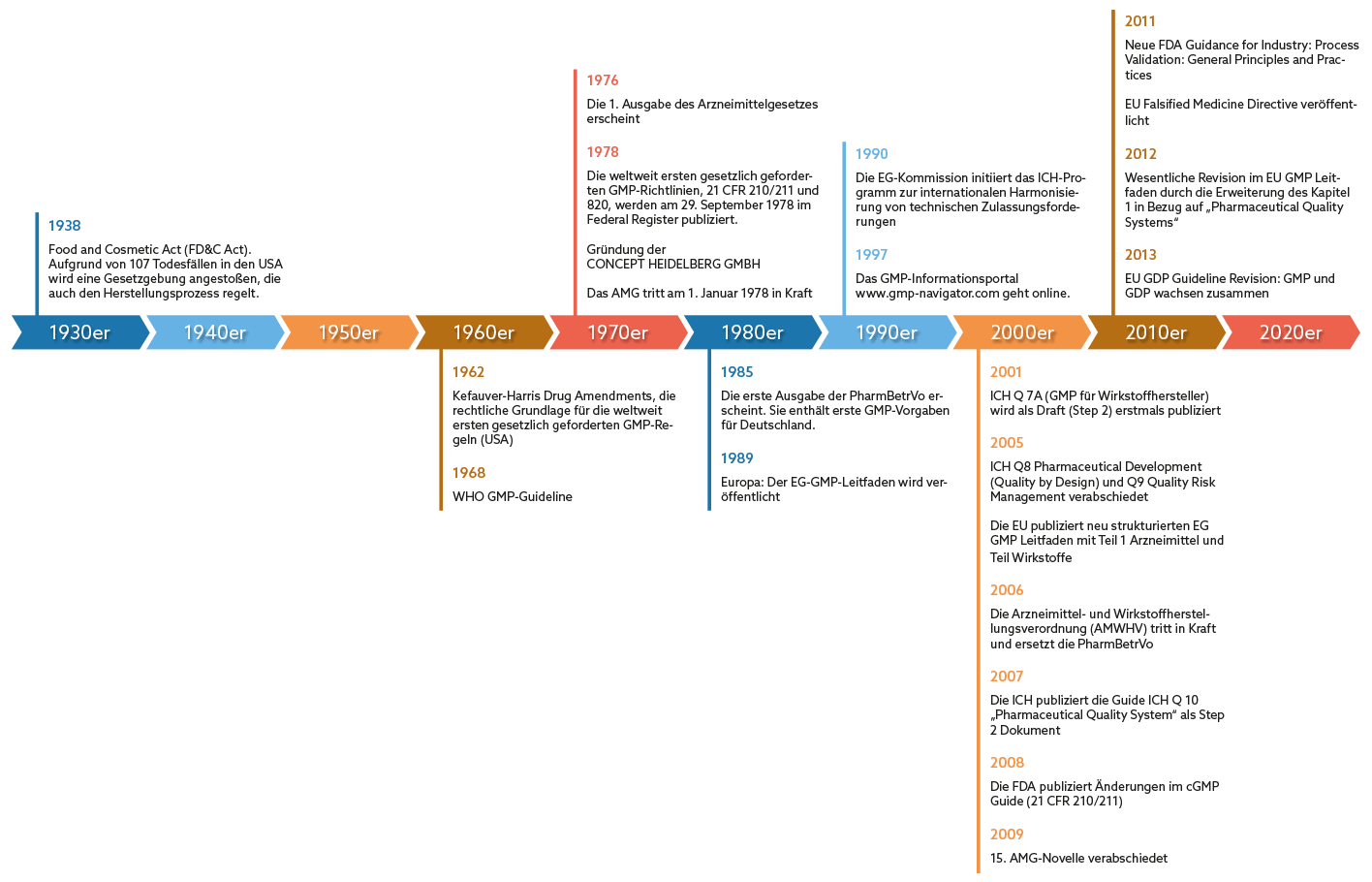
Grundlage für die GMP-Anforderungen ist der cGMP (Current Good Manufacturing Practice) Guide, den die amerikanische Food & Drug Administration (FDA) 1978 im Federal Register publiziert hat. Damit hatte die Behörde die weltweit ersten gesetzlich verankerten GMP-Regularien geschaffen. Diese ermöglichten eine Überwachung der Herstellungsprozesse der Pharmaunternehmen durch eine Kontrollbehörde.
Doch auch international gewann das Thema an Bedeutung: In Deutschland wurde als direkte Folge des Contergan-Skandals im Jahr 1978 das Arzneimittelgesetz (AMG) veröffentlicht, in dem es jedoch noch keine konkreten GMP-Vorgaben gab. Erst 1985 ist die Pharma-Betriebsverordnung erschienen. Darin waren erstmals für Deutschland konkrete GMP-Vorgaben enthalten, die die Grundlage für die GMP-Überwachung durch Inspektionen der zuständigen Behörden bildeten. Es dauerte weitere vier Jahre, bis dann schließlich im Jahre 1989 der für Europa verbindliche EG-GMP-Leitfaden erschienen ist. Laut AMWHV §2 heißt der Leitfaden heute EU-GMP-Leitfaden und ist seit 2005 in Deutschland rechtsgültig, was bedeutet, dass die Einhaltung seiner Vorgaben belegt werden muss.
Häufig gestellte Fragen - Inhaltsverzeichnis
Für was steht GMP?
GMP steht für Good Manufacturing Practice und wird mit Guter Herstellungspraxis ins Deutsche übersetzt. Arzneimittel- und Wirkstoffhersteller müssen bei der Herstellung, Verarbeitung, Prüfung, Verpackung und Lagerung die GMP-Richtlinien einhalten. Damit soll sichergestellt werden, dass sich Patientinnen und Patienten darauf verlassen können, dass Medikamente immer die gleichen, höchsten Qualitätsansprüche erfüllen.
Was sind GMP-Regeln?
Die GMP-Regeln sind die Forderungen an die Qualitätssicherung der Herstellungsabläufe und -umgebung, die die Europäische Kommission in den Grundsätzen und Leitlinien der Guten Herstellungspraxis (GMP) für Humanarzneimittel und Wirkstoffe formuliert hat. Wie diese Regeln auszulegen sind, beschreibt der EU-GMP-Leitfaden in Teil I für Arzneimittel und in Teil II für Wirkstoffe sowie in diversen Anhängen für bestimmte Produktgruppen.
(Quelle: https://www.bundesgesundheitsministerium.de/gmp.html)
Wo wird GMP angewendet?
GMP gilt sowohl in der Pharma- als auch in der Kosmetik- und der Lebensmittelindustrie. Neben Arzneimittel-Herstellern müssen also auch Hersteller von Kosmetika und von Lebensmitteln die Grundsätze der Guten Herstellungspraxis in Produktion, Qualitätssicherung und -kontrolle anwenden und damit sicherstellen, dass ihre Materialien und Produkte für den Endverbraucher sicher sind. Es ist jedoch wichtig zu betonen, dass die Anforderungen für die genannten Branchen unterschiedlich sind. Für Medikamente gelten besonders strenge Regeln zur Gewährleistung der Patientensicherheit.

Wo sind die GMP-Anforderungen definiert?
Die GMP-Vorgaben hat die Europäische Kommission im EU-GMP-Leitfaden zusammengefasst, der 1989 in Europa veröffentlicht wurde. Teil 1 des Leitfadens deckt dabei die Arzneimittelherstellung ab, Teil 2 die Herstellung von Wirkstoffen. Weitere 19 Anhänge (Annexe) richten sich an bestimmte Produktgruppen (wie z.B. sterile Arzneimittel, biologische Arzneimittel) oder enthalten weitere Vorgaben, wie z.B. für computergestützte Systeme).
Gilt GMP auch für Wirkstoffe?
Ja, auch für Unternehmen, die Wirkstoffe herstellen, die in Medikamenten verarbeitet werden, gilt GMP. Und auch für diese Wirkstoffbetriebe ist eine GMP-Überwachung vorgesehen. Allerdings ist die Durchführung einer GMP-Inspektion nicht Voraussetzung für die Erteilung einer Herstellungserlaubnis wie bei Arzneimitteln. Bei Wirkstoffen entscheidet die Behörde selbst, wann eine Inspektion durchzuführen ist. Für einige Wirkstoffe gilt allerdings eine Ausnahme, so z. B. Wirkstoffe, die tierischer, menschlicher oder mikrobieller Herkunft sind oder auf gentechnischem Wege hergestellt werden. In diesem Fall bedarf es einer Herstellungserlaubnis nach AMG §13 bzw. bei der Einfuhr eines Zertifikats nach AMG §72a(1)2. Die Überwachung erfolgt allerdings analog.
Wo stehen die GMP-Anforderungen an Wirkstoffhersteller?
Die GMP-Forderungen an Hersteller von Wirkstoffen sind ebenfalls im EU-GMP-Leitfaden definiert. Der Leitfaden ist in zwei Teile gegliedert. Teil I regelt die Arzneimittelherstellung, Teil II die Herstellung von Wirkstoffen.
Was ist eine GMP-Leitlinie?
Eine Leitlinie (Guideline) wendet sich an Fachkreise und ist primär erst einmal rechtlich nicht verbindlich. Begründete Abweichungen von den festgelegten Erwartungen sind möglich. Bestes Beispiel hierfür ist der EU-GMP-Leitfaden. Auf diesen Leitfaden wird allerdings in der deutschen Arzneimittel- und Wirkstoffherstellungsverordnung (AMWHV) verwiesen, die eine Durchführungsverordnung des Arzneimittelgesetzes (AMG) in Deutschland ist: "Zur Auslegung der Grundsätze der Guten Herstellungspraxis gilt für Arzneimittel, Blutprodukte im Sinne von § 2 Nr. 3 des Transfusionsgesetzes und andere Blutbestandteile sowie für Produkte menschlicher Herkunft der Teil I und für Wirkstoffe der Teil II des EU-GMP-Leitfadens." (§3(2)).
Was ist der Unterschied zwischen GMP und cGMP?
In den USA bezieht man sich häufig statt auf GMP auch cGMP (oder auch CGMP). Dabei steht das „C“ im für „current“. Die FDA sieht es letztendlich als Verantwortung der Industrie, mit Hilfe von Guides und Guidances, Inspectional Guidelines, Warning Letters, FDA Präsentationen und Workshops usw. auf dem Laufenden zu bleiben („current“). Daher auch die Begrifflichkeit des cGMP („current GMP“). Die Erwartungen der EU sind deutlicher zu erfassen.

Wer hat GMP entwickelt?
1962 bildeten dann die Kefauver-Harris Drug Amendments die rechtliche Grundlage für die weltweit ersten gesetzlich geforderten GMP-Regeln in den USA. Allerdings gab es noch keine konkreten Guidelines mit Umsetzungsempfehlungen. Die WHO veröffentlichte jedoch im gleichen Jahr die ersten GMP Guidelines. Diese waren aber rechtlich ohne Bedeutung, denn es bestand keine gesetzliche Pflicht, die GMP-Vorgaben bei der Herstellung von Arzneien umzusetzen. Im Jahre 1978 wurde schließlich der cGMP Guide der amerikanischen Food & Drug Administration (FDA) im Federal Register publiziert. Damit hatte die Behörde die weltweit ersten gesetzlich verankerten GMP-Regularien geschaffen. Diese ermöglichten eine Überwachung der Herstellungsprozesse der Pharmaunternehmen durch eine Kontrollbehörde.
In Deutschland wurde im Jahr 1978 das Arzneimittelgesetz (AMG) veröffentlicht, es gab darin jedoch keine konkreten GMP-Vorgaben. Das AMG war eine direkte Folge des Contergan-Skandals. Erst 1985 ist die Pharma Betriebsverordnung erschienen. Darin waren erstmals für Deutschland konkrete GMP Vorgaben enthalten, diese bildeten die Grundlage für die GMP-Überwachung durch Inspektionen der zuständigen Behörden. Es dauerte weitere 4 Jahre, bis dann schließlich im Jahre 1989 der für Europa verbindliche EG GMP Leitfaden erschienen ist.
Ist GMP ein Gesetz?
GMP selbst ist nicht gesetzlich. Im GMP-Bereich in Deutschland ist für Arzneimittel- und Wirkstoffhersteller v. a. das Bundesrecht von Interesse und hier insbesondere das AMG (Arzneimittelgesetz) und die AMWHV (Arzneimittel- und Wirkstoffherstellungsverordnung), die sich in §3(2) auf den EU-GMP-Leitfaden bezieht. Damit wird die Einhaltung der GMP-Anforderungen für beide verbindlich. Zusätzlich gibt es noch eine Reihe an EU-Verordnungen („Regulations“), die direkt bindend wirksam sind.
Ist GMP Pflicht?
GMP ist für alle Hersteller verpflichtend, die ein Medikament zulassen und auf den Markt bringen wollen. Dabei gilt jedoch, dass die GMP-Richtlinien des Landes anzuwenden sind, in dem der Standort für dessen Herstellung liegt. Für ein Unternehmen mit Arzneimittelproduktion in Deutschland ist z. B. der EU-GMP-Leitfaden ausschlaggebend. Die darin definierten GMP-Anforderungen werden durch das deutsche Arzneimittelgesetz (AMG) und die Arzneimittel- und Wirkstoffherstellungsverordnung (AMWHV) als Durchführungsverordnung verpflichtend. Führt das Unternehmen seine Produkte jedoch auch in die USA aus, ist auch der US FDA GMP Guide anzuwenden. Sonst könnte es Probleme bei Zulassungen oder bei der Einfuhr der Produkte in die USA geben.
Wer überprüft die Einhaltung von GMP?
Die Einhaltung wird durch die staatlichen Arzneimittelbehörden (GMP/GDP Inspektorate) überwacht. In Deutschland erfolgt die Überwachung z. B. in Verantwortung der Bundesländer. In der Regel ist das Regierungspräsidium oder die Bezirksregierung dafür zuständig. Dabei ist der Produktionsstandort für ein Medikament entscheidend. Ein Produktionsstandort in Frankfurt wird beispielsweise vom Regierungspräsidium Darmstadt überwacht. Auf der Website der „Zentralstelle der Länder für Gesundheitsschutz bei Arzneimitteln und Medizinprodukten“ (ZLG) ist eine Liste der zuständigen Länderbehörden zu finden.
Welche GMP-Überwachungsstellen gibt es in Deutschland?
Insgesamt gibt es 37 Überwachungsbehörden, darunter 27 GMP-Inspektorate, welche die Überwachung von Herstellern und Einführern durchführen. Es gibt sechs Inspektorate für Tierimpfstoffhersteller und zusätzlich zehn Arzneimitteluntersuchungsstellen. Eine Liste der Überwachungsbehörden in Deutschland ist auf der Website der ZLG (Zentralstelle der Länder für Gesundheitsschutz bei Arzneimitteln und Medizinprodukten) abrufbar.
Wer führt GMP-Inspektionen durch?
Eine GMP-Inspektion im Pharma-Umfeld kann ausschließlich eine staatliche Behörde durchführen. Akkreditierte Stellen, die z.B. ISO-Audits durchführen, dürfen keine offiziellen GMP-Inspektionen veranlassen oder die GMP-Konformität zertifizieren. Es gibt es auch keine GMP-Zertifizierung / kein GMP-Zertifikat der Behörde, wohl aber eine Bescheinigung. Die offizielle Namensgebung ist: „Bestätigung der Übereinstimmung eines Herstellers mit GMP“. Umgangssprachlich wird das oft mit „GMP-Zertifikat“ bezeichnet. Die Grundlage für die Durchführung bildet die EU GMP-Richtlinie 2001/83/EG. Diese Richtlinie wurde in deutsches Recht umgesetzt. Die Behörde inspiziert auf Grundlage der Anforderungen in der EU Richtlinie 2003/94 EG. Die konkrete Auslegung findet sich im EU-GMP-Leitfaden inklusive der Anhänge. In Deutschland kommt für die Umsetzung auch die Arzneimittel- und Wirkstoffherstellungsverordnung zur Anwendung. Diese wurde erlassen, um eine konkrete Durchführungsverordnung zu haben. Diese Durchführungsverordnung enthält z.B. auch in § 42 den Ordnungswidrigkeitenkatalog für den Fall, dass Unternehmen gegen die GMP-Vorgaben verstoßen.
Was ist ein GMP-Zertifikat?
Anders als bei der ISO-Norm werden Hersteller von Arzneimitteln nicht „GMP-zertifiziert“. Allerdings kann die zuständige Überwachungsbehörde einem Betrieb, der eine Herstellungserlaubnis besitzt – also nach GMP arbeitet und überwacht wird – auf Antrag des Herstellers ein WHO GMP-Zertifikat ausstellen. Arzneimittelhersteller benötigen diese Zertifikate oft für den Export, da der Zoll von nicht EU-Ländern diese Zertifikate in der Regel anfordert. Daher werden WHO-Zertifikate oft auch als Exportzertifikate bezeichnet.
Auch Firmen, die z. B. Anlagen für die pharmazeutische Produktion entwickeln oder andere herstellungsnahe Tätigkeiten durchführen, erhalten kein GMP-Zertifikat. Der pharmazeutische Unternehmer ist dafür verantwortlich, dass er Dienstleister und Zulieferer auswählt, die GMP-gerechte Leistungen anbieten. Im Rahmen einer Lieferantenqualifizierung muss er nachweisen, dass er die ausgewählten Lieferanten initial und dann regelmäßig überwacht. Dafür muss der pharmazeutische Unternehmer einen risikobasierten Ansatz wählen. Bei Lieferanten, deren Produkte ein geringes potentielles Risiko für die Patientensicherheit aufweisen, kann die Überprüfung z. B. über Selbstbewertungsbögen erfolgen. Bei Dienstleistungen, die potentiell ein hohes Risiko für die Patientensicherheit bergen, mag eine GMP-Überprüfung des Lieferanten durch ein GMP-Audit erforderlich sein.
Was ist GMP zertifiziert?
Den Begriff „GMP-zertifiziert“ gibt es an sich nicht. Trotzdem wird „GMP-Zertifikat“ häufig für die „Bestätigung der Übereinstimmung eines Herstellers mit GMP“ verwendet, die die offizielle Behörde ausstellt. Hier finden Sie mehr zum Thema GMP Zertifizierung und GMP Zertifikat.
Was ist eine GMP-Qualifizierung?
Sowohl der EU-GMP-Leitfaden als auch der cGMP Guide der US-amerikanischen FDA fordern, dass Hersteller über Personal in ausreichender Zahl und mit der erforderlichen Qualifizierung und praktischen Erfahrung verfügen. Mitarbeiter/innen, die in der Herstellung, Prüfung und Lagerung von Arzneimitteln oder Wirkstoffen arbeiten, müssen also für Ihre jeweiligen Aufgaben qualifiziert und fortlaufend weitergebildet sein/werden. Jedes pharmazeutische Unternehmen muss aus diesem Grund ein umfassendes Schulungssystem etablieren, welches GMP-Schulungen plant und dokumentiert.
Was ist ein GMP-Audit?
GMP-Audits sind in der pharmazeutischen Industrie ein zentrales Element zur Umsetzung qualitätssichernder Maßnahmen. Durch ein Audit können Schwachpunkte aufgezeigt und beseitigt und damit sichergestellt werden, dass das entsprechende Unternehmen daran arbeitet, die Qualität und Sicherheit seiner Produkte zu gewährleisten. Laut AMWHV §11 (Selbstinspektionen und Lieferantenqualifizierung), Absatz 3 ist ein Audit „die Durchführung von Überprüfungen des Herstellers vor Ort“.
Was ist GMP-gerechte Dokumentation?
Unter einer GMP-gerechten oder GMP-konformen Dokumentation versteht man zum einen Dokumente, die Mitarbeiter:innen anweisen, wie sie bestimmte Prozesse auszuführen haben – beispielsweise Standard Operating Procedures (SOPs) oder Verfahrensanweisungen – und zum anderen Dokumente, die gegenüber den prüfenden Behörden belegen, dass die in den SOPs oder Verfahrensanweisungen festgelegten Schritte befolgt wurden, wie etwa Protokolle oder Berichte.
Was ist eine Herstellungs- und/oder Einfuhrerlaubnis?
Pharmazeutische Hersteller in der EU, die Human-, Tier- oder Prüfarzneimitteln herstellen und/oder aus einem Drittland einführen, benötigen eine Herstellungs-/Einfuhrgenehmigung (Manufacturing and Importation Authorisation; MIA). Wenn das Unternehmen rein am Warenverkehr innerhalb des EWR beteiligt ist, ist nur eine Großhandelsvertriebsgenehmigung erforderlich.
Siehe auch Gesetz über den Verkehr mit Arzneimitteln (Arzneimittelgesetz - AMG), §13, Herstellungserlaubnis: „Wer
- Arzneimittel,
- (weggefallen)
- Wirkstoffe, die menschlicher, tierischer oder mikrobieller Herkunft sind oder die auf gentechnischem Wege hergestellt werden, oder
- andere zur Arzneimittelherstellung bestimmte Stoffe menschlicher Herkunft
gewerbs- oder berufsmäßig herstellt, bedarf einer Erlaubnis der zuständigen Behörde.“
Und §72 Einfuhrerlaubnis: (1) „Wer
- Arzneimittel,
- Wirkstoffe, die menschlicher, tierischer oder mikrobieller Herkunft sind oder die auf gentechnischem Wege hergestellt werden, oder
- andere zur Arzneimittelherstellung bestimmte Stoffe menschlicher Herkunft
gewerbs- oder berufsmäßig aus Ländern, die nicht Mitgliedstaaten der Europäischen Union oder andere Vertragsstaaten des Abkommens über den Europäischen Wirtschaftsraum sind, in den Geltungsbereich dieses Gesetzes einführen will, bedarf einer Erlaubnis der zuständigen Behörde.“
Diese Genehmigung erlaubt es also Pharmaunternehmen, Arzneimittel in der EU herzustellen und/oder zu importieren.
Die Erlaubnis wird pharmazeutischen Herstellern wird in der Regel von den zuständigen nationalen Behörden oder von der EMA erteilt, je nach Art des Arzneimittels und des Zulassungsverfahrens. Die Einfuhrgenehmigung wird erteilt, nachdem überprüft wurde, dass die importierten Produkte die gleichen GMP-Standards erfüllen, die für in der EU hergestellte Produkte gelten.
Die Einhaltung der EU-GMP-Standards ist für die Aufrechterhaltung dieser Zulassungen unerlässlich. Die Aufsichtsbehörden führen regelmäßig Inspektionen durch, um die laufende Einhaltung der Standards zu überprüfen, und die Nichteinhaltung der Standards kann zum Widerruf der Genehmigung und zu anderen behördlichen Maßnahmen führen.
Was ist GDP?
Unter Good Distribution Practice (GDP) versteht man die Summe der Maßnahmen, die durch eine Kontrolle der Vertriebskette sicherstellt, dass die Qualität und die Unversehrtheit von Arzneimitteln aufrechterhalten wird. Zwar richtet sich die EU GDP-Leitlinie primär an den Arzneimittelgroßhandel, aber die Umsetzung der Vorgaben muss gleichermaßen durch alle Beteiligten in der Vertriebskette sichergestellt werden. Daher müssen auch Arzneimittelhersteller und jegliche Beteiligte in der Logistik (Transportfirmen, Lager, etc.) die GDP-Anforderungen erfüllen, sofern diese im Rahmen der Arzneimitteldistribution involviert sind. Für pharmazeutische Wirkstoffe hat die EU-Kommission ebenfalls eine Leitlinie formuliert. Diese trägt den Titel „Grundsätze der guten Vertriebspraxis für Wirkstoffe von Humanarzneimitteln“.
Die GDP-Anforderungen werden von den GMP-/GDP-Inspektoraten der einzelnen Länder überwacht. Im Rahmen von GDP-Inspektionen wird geprüft, ob sie entsprechend den Vorgaben der Guideline in die betriebliche Praxis umgesetzt wurden. Dazu werden die Räumlichkeiten, Prozesse und vor allem die Vorgabe- und Nachweisdokumentation regelmäßig überprüft.
Was ist die GMP-Richtlinie?
Mit der GMP-Richtlinie 2017/1572 der Europäischen Union (EU) möchte die EU GMP-Anforderungen an die Hersteller von Arzneimitteln in allen EU-Mitgliedsstaaten vereinheitlichen. EU-Richtlinien müssen allerdings in den einzelnen Mitgliedsstaaten in nationales Recht umgesetzt werden. Dies erfolgte z. B. in Deutschland über die AMWHV und in Österreich über die AMBO. Die EU-GMP-Richtlinie ist mit sieben Seiten relativ „schlank“, gibt aber mit ihrer Gliederung in
- Pharmazeutisches Qualitätssystem
- Personal
- Räumlichkeiten und Ausrüstung
- Dokumentation
- Herstellung
- Qualitätskontrolle
- Auftragsherstellung
- Beanstandungen und Produktrückruf
- Selbstinspektionen
schon den Rahmen für die pharmazeutischen Hersteller vor. Neben der Umsetzung in z. B. die AMWHV gibt auch der EU-GMP-Leitfaden mit seinen Anhängen Interpretationshilfe zu diesen insgesamt neun o. g. Paragraphen.
Seminare und Schulungen
Für Hersteller von Arzneimitteln oder Wirkstoffen bedeutet das, dass sie ihre Mitarbeiter für die GMP-konforme Durchführung ihrer Aufgaben angemessen schulen müssen. Dieses Training soll dazu dienen, dass Produktionsmitarbeiter die für ihren Einsatzbereich wichtigen GMP-Vorgaben kennen lernen und nach erfolgreichem Abschluss des Kurses umzusetzen wissen. Für diese Schulung muss außerdem ein Nachweis erbracht werden. Dabei unterstützt Concept Heidelberg Firmen weltweit und bietet ein breites Spektrum an GMP-Seminaren, GMP-Lehrgängen, Weiterbildungen und Konferenzen an. Angefangen bei den GMP-Grundlagen und bis ins Detail vermitteln wir GMP-Kenntnisse in Bezug auf Produktion, Dokumentation, Qualitätsmanagement, Mikrobiologie, Aseptik, Lagerung usw. Seit einiger Zeit bieten wir unsere Seminare außerdem auch als Online-Seminare an. Informieren Sie sich über unsere Seminare, welche Sie auch unter dem Menüpunkt Seminare finden.

